SINDROME DI KLINEFELTER
La Sindrome di Klinefelter è una malattia genetica che si manifesta soprattutto con alterazione della differenziazione sessuale e insufficienza mentale. Il nome è stato dato in onore di Harry Klinefelter che descrisse la malattia per la prima volta nel 1942, per la verità lavorando assieme a Fuller Albright.
EPIDEMIOLOGIA
Si calcola che ne siano affetti circa 1-2 maschi ogni 1000 nati vivi: non sembra esistano etnie in cui sia più o meno frequente.
EZIOLOGIA E PATOGENESI
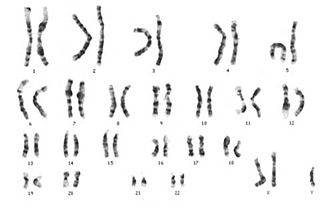
Sindrome di Klinefelter: cariotipo 47,XXY
fonte: Wikipedia.org
L’anomalia genetica che venne descritta da Klinefelter è una alterazione del cariotipo caratterizzata da un cromosoma X sovrannumerario; il cariotipo classicamente viene definito definito come “47, XXY”. In realtà successivamente venne dimostrato che lo stesso quadro clinico può essere provocato da alterazioni cariotipiche diverse o anche da alcuni mosaicismi; 48, XXYY; 48, XXXY; 49XXXYY; XXY/XY; XXY/XX; XXY/XXXYY.
Le forme più frequenti sono la 47,XXY e il mosaicismo XXY/XY che coprono rispettivamente l’80% e il 10% dei casi.
IL meccanismo che provoca la presenza del crmosoma soprannumerario non è stato ancora accertato ma la teoria maggiormente accreditata è che durante la meiosi, cioè durante il processo di scissione dei cromosomi che avviene durante la maturazione degli spermatozoi e degli ovuli, si crea una condizione per cui uno dei gameti mantiene la coppia originaria, quindi XX (donna) o XY (uomo).
Il gamete XX proveniente dalla madre, se si fonde con un gamete Y proveniente dal padre produce un individuo XXY, quindi con sindrome di Klinefelter.
Il gamete XY proveniente dal padre, se si fonde con un gamete X proveniente dalla madre produce un individuo XXY, anche in questo caso con sindrome di Klinefelter.
Il gamete XX proveniente dalla madre, se si fonde con un gamete X proveniente dal padre produce un individuo XXX, fenotipicamente donna, con una sindrome chiamata del “Triplo X”, differente dalla Klinefelter.
L’alterazione cromosomica XXY, per una serie di complessi meccanismi relativi all’espressione del gene per il testoterone, provoca alterazioni caratteristiche della funzione delle gonadi e altre alterazioni fenotipiche.
QUADRO CLINICO.
I soggetti affetti, fenotipicamente maschi, in periodo prepuberale, e comunque in una minoranza dei casi, possono manifestare insufficienza mentale e brevità dei metacarpi, ma spesso l’unico segno evidenziabile è una lieve ipoplasia testicolare; non raro però un ritardo nello sviluppo del linguaggio.
Alla pubertà si instaura un quadro clinico caratterizzato da testicoli di volume molto ridotto e con consistenza aumentata, da comparsa di ginecomastia, da quello che viene chiamato aspetto “eunucoide” con arti sproporzionatamente lunghi rispetto al tronco, ridotta crescita dei peli della barba e dei peli di pube e arti-tronco, iposviluppo della muscolatura scheletrica e dei genitali esterni. Il deficit intellettivo non è molto frequente, ma le turbe del linguaggio persistono anche in epoca adulta.
Di solito i soggetti con Sindrome di Klinefelter raggiungono una statura maggiore dei genitori. In epoca adulta è segnalata una maggiore incidenza di diabete, di osteoporosi, di cardiopatie, di distiroidismi e di alcuni tumori e in particolare tumori mammari, polmonari e linfomi.
Nei mosaicismi il quadro clinico può essere più sfumato, a volte così impercettibile da essere scoperto solo dopo esami per l’infertilià tipica di questi soggetti. Viceversa, nelle forme con più di un cromosoma X sovrannumerario, il quadro clinico da ipogonadismo è più severo e i deficit intellettivi sono più pronunciati.
DIAGNOSI.
La diagnosi, oltre che sul quadro clinico che fornisce il sospetto, è confermata dall’analisi genetica del cariotipo.
Il laboratorio, oltre all’alterazione genetica, documenta, in epoca post-puberale, elevati livelli di FSH ed LH, bassi livelli di testosterone, normali o poco aumentati livelli di 17-β-estradiolo, aumentati livelli di prolattina. Costante è l’azoospermia.
La biopsia testicolare può documentare fibrosclerosi dei tubuli seminiferi con cellule germinale assenti o gravemente ridotte.
TERAPIA
L’unica terapia possibile è sostitutiva del deficit androgenico, quindi si utilizza testosterone a partire dalla pubertà.
L’azospermia in via teorica non è passibile di trattamento, ma in letteratura sono segnalati numerosi casi di gravidanze ottenute con tecniche di procreazione assistita.
Testi consigliati:
Principi di medicina interna 20 ed. Copertina rigida – 11 gennaio 2021 di Harrison
AMAZON: principi di medicina interna XX edizione, copertina rigida- Harrison
Dott. Salvatore Nicolosi